
In patients with anti-PM/Scl autoantibodies, which target components of the nuclear RNA exosome complex, RNAseq showed over-expression of 236 RNAs, mostly long noncoding or divergent transcripts, suggestive of dysfunction of the nuclear RNA exosome complex and correlated with transcriptomic disease activity markers.7
Autoantibodies as more than biomarkers
Historically, autoantibodies in AIM were viewed primarily as diagnostic or classification tools. However, accumulating evidence supports a more active role in disease pathophysiology.4,5
Distinct autoantibody profiles are associated with characteristic patterns of muscle injury, extramuscular organ involvement, and inflammatory response, suggesting that autoantibodies may contribute directly to disease mechanisms rather than simply reflecting immune activation.1,2

In AIM, immunoglobulin G (IgG) autoantibodies, may be central pathological factors driving the disease3,6,7
Recent data challenge the assumption that myositis autoantibodies against intracellular antigens are purely epiphenomenal, showing that these autoantibodies can be internalized into muscle cells and disrupt cellular pathways3,7

The different subclasses and modes of action of IgG autoantibodies collectively determine the disease phenotype and target organ damage.6
Pathogenicity of autoantibodies
In autoimmune myositis, autoantibodies appear to act upstream of Type I interferon amplification rather than simply marking downstream inflammation.7
669 muscle biopsy samples were isolated from patients with myositis, disease controls, and healthy controls. Bulk RNAseq analysis was performed, with a subset of samples undergoing immunofluorescence staining.7
IgG immunofluorescence staining revealed cytoplasmic deposition of antibodies in myofibres of patients with anti-HMGCR, anti-SRP, anti-MDA5 and anti-Jo1 autoantibodies, all of which target autoantigens in the cytoplasm.7
Anti-Mi2 autoantibodies, which recognize a transcription regulator, the Mi2/NuRD complex, induced de-repression of 100 genes associated with disease activity; similar findings were observed when purified autoantibodies from anti-Mi2+ dermatomyositis (DM) patients were internalized to cultured muscle cells.7
These data show that autoantibodies find their way inside muscle cells where they disrupt the function of their targets, providing mechanistic evidence that myositis autoantibodies are pathogenic.7

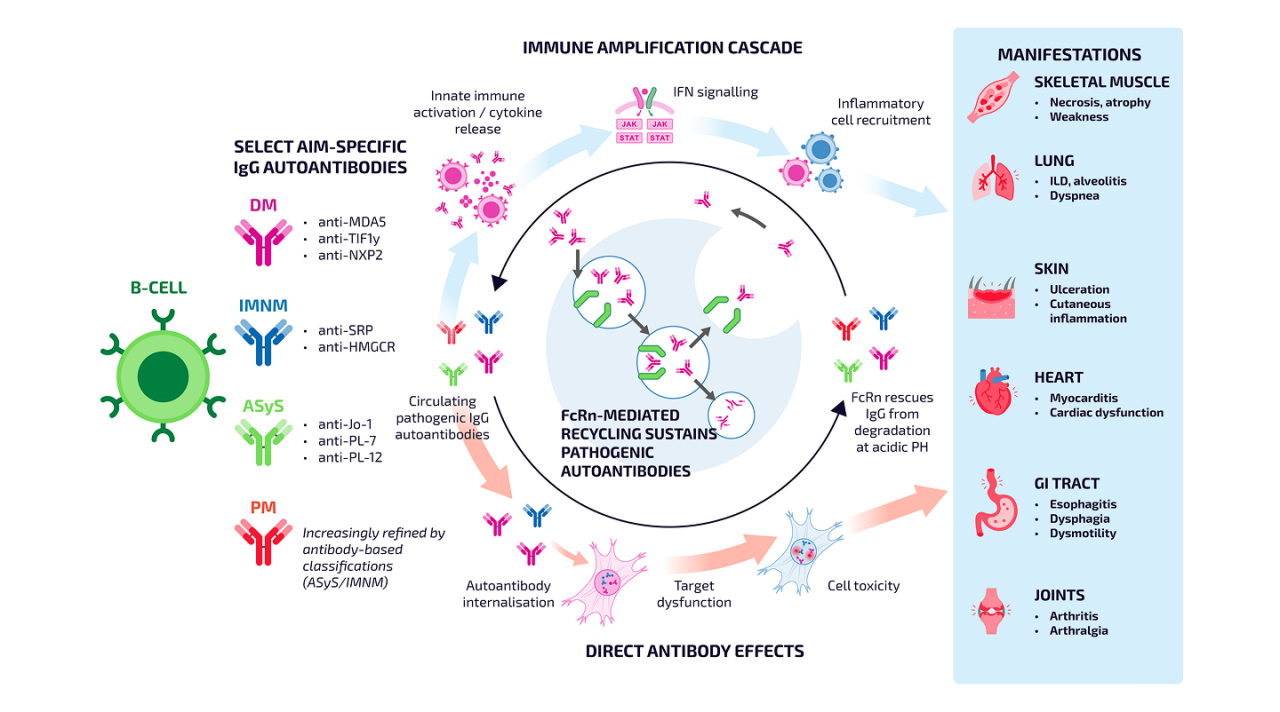
Neonatal Fc receptor (FcRn) and the persistence of pathogenic autoantibodies
The FcRn plays an important role in regulating IgG homeostasis by protecting IgG antibodies from intracellular degradation and extending their circulating half-life.6
In AIM, this recycling pathway may contribute to the persistence of pathogenic IgG autoantibodies associated with ongoing immune activation and tissue injury. By rescuing IgG from lysosomal degradation and returning it to circulation, FcRn helps maintain circulating autoantibody levels.6
Increasing understanding of FcRn-mediated IgG recycling has highlighted its relevance to autoantibody-driven diseases, including AIM, and has contributed to interest in therapeutic approaches targeting this pathway.6
FcRn-mediated persistence of pathogenic AIM autoantibodies may drive immune amplification and tissue injury5–15
When an IgG antibody connects to a FcRn receptor, it gets recycled back into the bloodstream and stays in circulation.6
Adapted from references 5–15
Continue exploring AIM
Learn more about the role of autoantibodies in AIM.
MSAs and MAAs
Identifying autoantibody profiles is transforming our understanding of AIM.1,2
Diagnosis of AIM
Autoantibody testing has become an increasingly important component of diagnostic evaluation and subtype classification.1,8
Abbreviations:
AAB, autoantibody; AIM, autoimmune myositis; ASyS, anti-synthetase syndrome; DM, dermatomyositis; FcRn, neonatal Fc receptor; HMGCR, 3-hydroxy-3-methylglutaryl-coenzyme A reductase; IFN, interferon; IgG, immunoglobulin G; ILD, interstitial lung disease; IMNM, immune-mediated necrotizing myopathy; JAK-STAT, Janus kinase–signal transducer and activator of transcription; MAA, myositis-associated autoantibody; MDA5, melanoma differentiation‑associated gene 5; MSA, myositis-associated autoantibody; NXP2, nuclear matrix protein 2; PL‑7, threonyl‑tRNA synthetase; RNA, ribonucleic acid; SRP, signal recognition particle; TIFIy, transcription intermediary factor 1-gamma.
References:
1. Wang G and McHugh NJ. Clin Exp Rheumatol. 2025;43(2):364-371; 2. Wu Y, et al. Front Immunol. 2024;15:1439807; 3. Musai J, et al. Curr Rheumatol Rep. 2024;26(12):421-430; 4. Wischnewski S, et al. Trens Pharmacol Sci. 2025;46(3):249-263; 5. Groener M and Paik J. Front Immunol. 2025;16:1581323; 6. Yang CW, et al. Front Immunol. 2025:16:1656937; 7. Pinal-Fernandez I, et al. Ann Rheum Dis. 2024;83(11):1549-1560; 8. Halilu F, Christopher-Stine L. Rheumatol Immunol Res. 2022;3:1–10; 9. Gasparotto M, et al. Autoimmun Rev. 2023 Jun;22(6):103334; 10. Martins EF, et al. Int J Mol Sci. 2025;26:3302; 11. Arouche-Delaperche L, et al. Ann Neurol. 2017;81(4):538–548; 12. Lundberg IE, et al. Nat Rev Dis Primers. 2021;7(1):86; 13. Saketkoo LA, et al. Curr Rheumatol Rev. 2010 May ; 6(2): 108–119; 14. Zhu H, et al. J Inflamm Res. 2025;18:3879–3888; 15. Ohmura SI, et al. Cureus. 2024;16(10):e71821